Gene expression analysis is one of the most powerful tools in modern biology. By examining which genes are turned on or off in a given tissue, researchers can uncover the molecular drivers of health, development, and disease. This knowledge informs everything from basic research in cell biology to the design of diagnostic tests and targeted therapies.
For nearly two decades, microarray technology was the dominant platform for transcriptome-wide analysis. With the advent of RNA sequencing (RNA-seq), powered by next-generation sequencing (NGS), many assumed that microarrays would soon be obsolete. After all, RNA-seq offers deeper resolution, detects novel transcripts, and reveals more complexity in the transcriptome.
Yet, the story is not so simple. When analyzed with consistent and robust statistical methods, results from microarrays and RNA-seq often show remarkable concordance. This suggests that both platforms remain valuable, particularly when combined with the right bioinformatics approaches.
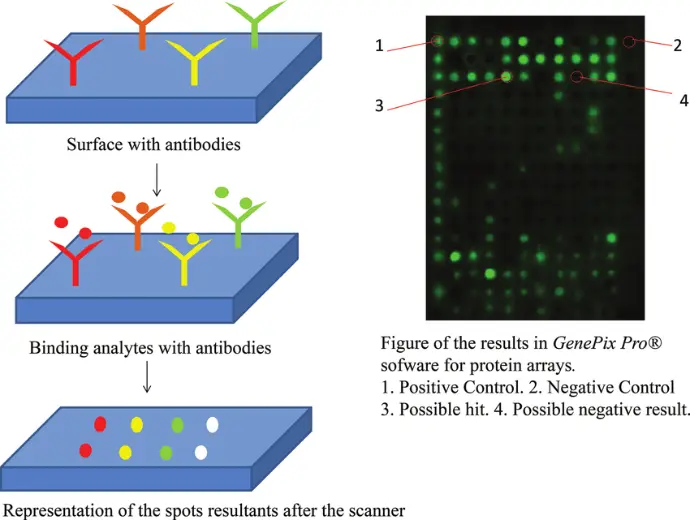
Microarrays: The Foundation of Transcriptomics
Microarrays were among the first technologies to make genome-wide expression profiling possible, providing researchers with the ability to study thousands of genes simultaneously.
How it works:
In a microarray experiment, RNA is extracted from a biological sample and converted into labeled cDNA. This cDNA is hybridized to a glass or silicon chip containing pre-designed DNA probes, each corresponding to a specific gene. The fluorescence intensity at each probe spot reflects the relative abundance of that gene’s transcript.
Key strengths:
High throughput: Microarrays can measure thousands of transcripts at once, enabling comprehensive profiling of the transcriptome.
Cost-effective: They are efficient for analyzing large numbers of samples, making them suitable for population-scale studies.
Reproducibility: Years of refinement have established highly standardized protocols, ensuring consistent and reliable results.
Rich legacy datasets: Public databases such as GEO (Gene Expression Omnibus) contain vast amounts of microarray data, allowing researchers to compare, validate, and integrate findings across studies.
Microarrays remain a cornerstone of gene expression research, especially in projects that benefit from affordability, scalability, and the ability to connect with decades of historical data.
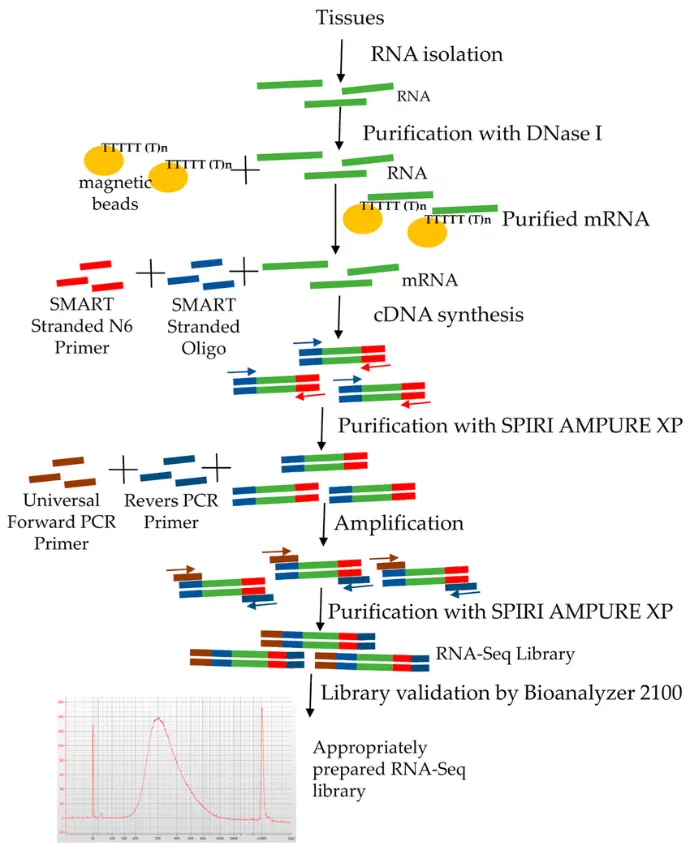
RNA-Seq: The Next-Generation Standard
With the rise of next-generation sequencing, RNA-seq brought a new level of detail and flexibility to transcriptomics. Unlike microarrays, RNA-seq does not rely on predefined probes it directly sequences transcripts, giving researchers a complete and dynamic view of the transcriptome.
How it works:
RNA from a sample is converted into cDNA, fragmented, and then sequenced on high-throughput platforms. The millions of short reads generated are aligned to a reference genome or assembled to reconstruct the entire transcriptome.
Key strengths:
Comprehensive coverage: RNA-seq can profile both known and novel transcripts, providing insights into protein-coding genes, isoforms, and non-coding RNAs.
High sensitivity: It can detect transcripts across a wide range of expression levels, including very rare transcripts.
Quantitative precision: By counting sequence reads, RNA-seq provides a direct measurement of gene abundance.
Multidimensional insights: Beyond expression levels, RNA-seq enables the study of alternative splicing, allele-specific expression, RNA editing, and transcript variation.
RNA-seq has become the method of choice for discovery-driven research, offering the depth and versatility needed for modern genomics.
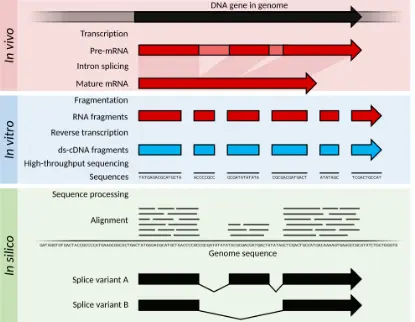
Together, microarrays and RNA-seq provide complementary strengths in transcriptomics research. Microarrays offer efficiency, reproducibility, and a wealth of historical data for comparative analysis, while RNA-seq delivers high-resolution, quantitative, and discovery-driven insights.
Comparing Outputs: Typical Observations
When applied to the same samples, microarrays and RNA-seq show:
High correlation in global gene expression patterns
Overlapping differentially expressed genes (DEGs), often hundreds of shared genes
Shared pathways identified in both platforms, reinforcing key biological insights
The choice of statistical approach plays a crucial role in achieving this concordance. Non-parametric methods, like the Mann–Whitney U test, improve agreement between platforms by accommodating the non-normal distribution of biological data.
Why Statistical Approaches Matter
The method used to analyze data can be as important as the technology itself. Non-parametric statistics allow researchers to:
Minimize bias caused by distribution assumptions
Identify robust DEGs across platforms
Enhance reproducibility and confidence in results
Practical Implications for Research
Complementary Use: Combining microarrays and RNA-seq leverages the strengths of both platforms.
Integration with Legacy Data: Decades of microarray datasets can be integrated with RNA-seq results to validate findings and uncover new insights.
Robust Biomarker Discovery: Genes and pathways identified by both platforms are more likely to represent true biological signals, critical for clinical research.
Recommended Product for RNA Extraction
High-quality RNA is essential for both microarray and RNA-seq workflows.
RNA Extraction Kit
- Efficiently isolates high-quality RNA from a wide range of biological samples
- Provides RNA suitable for cDNA synthesis, microarrays, or RNA-seq library preparation
- Optimized for reproducibility and high yield